
Vanqua Bio harnesses leading-edge knowledge of lysosomal biology and the innate immune system, a screening platform using patient iPSC-derived neuronal cells, along with proven drug discovery and development expertise, to discover novel disease-modifying therapies. Our approach will deliver first-in-class and best-in-class therapies to slow or stop the progression of PD, AD, and related neurological diseases.
Our Pipeline

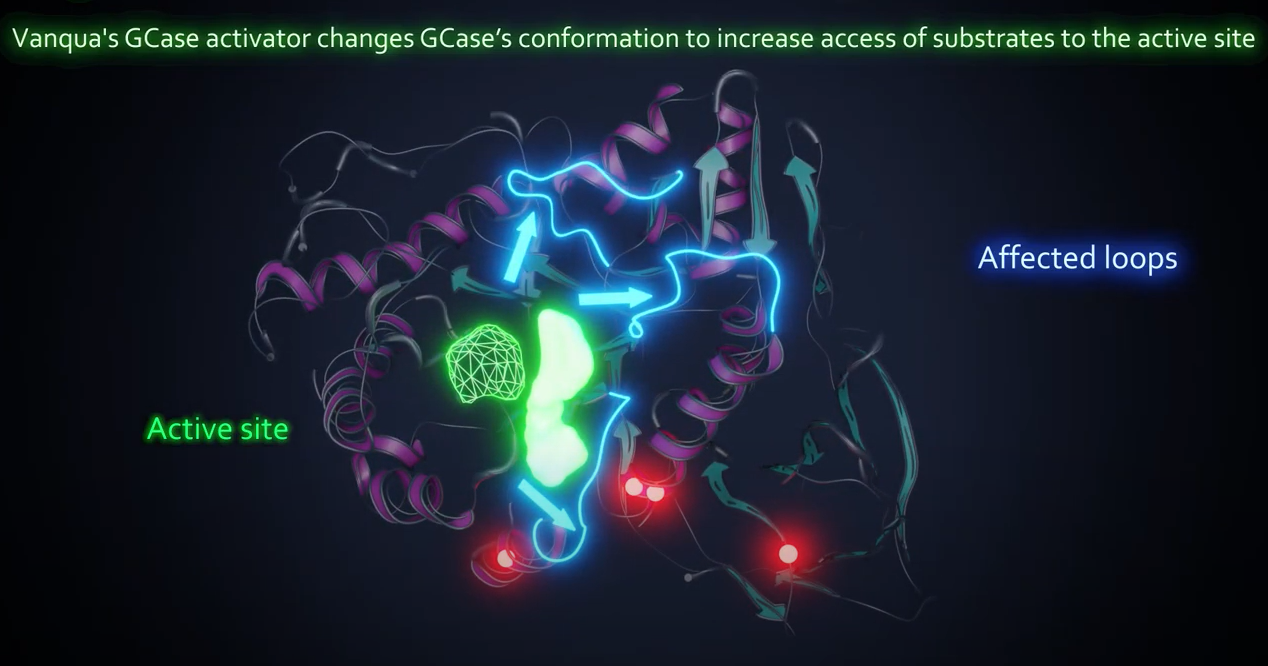
GCASE – GBA Parkinson’s Disease
Vanqua Bio’s Scientific Focus: Our lead program targets the genetic association between the GBA1 gene, the gene that encodes GCase, and the lysosomal dysfunction that occurs in Parkinson’s disease (GBA-PD).
Mutations in GBA1 are the most common genetic risk factor for PD. Roughly 10% of patients with PD in the U.S. and Europe carry a GBA1 mutation. Individuals harboring GBA1 mutations often develop PD at a younger age and exhibit more rapid disease progression compared to patients with idiopathic PD.
In patients with PD, mutations in one copy of the GBA1 gene can decrease GCase activity by up to 50%. The magnitude of decrease in enzymatic activity is strongly associated with the severity of symptoms. Decreased GCase activity is also observed in idiopathic PD (PD that occurs in patients without GBA1 mutations) and Lewy Body Dementia.
Functional GCase is crucial for the recycling and disposal of proteins and lipids in the lysosome. Numerous scientific studies have demonstrated that GCase mutations trigger lysosomal dysfunction, cell toxicity, inflammation, and the accumulation of alpha synuclein (a hallmark of PD), which is toxic to neurons.
GBA mutations trigger a cascade of negative effects in neuronal cells, leading to Parkinson’s Disease

Vanqua Bio Therapeutic Hypothesis: Small molecule activators of GCase will restore lysosomal function and attenuate accumulation of alpha synuclein and neurodegeneration.

GCASE – Idiopathic Parkinson’s
Idiopathic Parkinson’s disease patients, despite not having a mutation in GBA1, can also have decreased GCase activity, albeit to a lesser degree than in GBA-PD patients. While GBA-PD patients progress more quickly than idiopathic PD, the two diseases share similar clinical presentation and pathology. As such, lysosomal dysfunction and the subsequent accumulation of alpha synuclein is also heavily implicated as a driver of neurodegeneration in idiopathic Parkinson’s.
Vanqua Bio Therapeutic Hypothesis: Small molecule activators of GCase will improve lysosomal function in idiopathic Parkinson’s patients and attenuate the accumulation of alpha synuclein and neurodegeneration.
GCASE – GBA Dementia with Lewy Bodies
Dementia with Lewy bodies (DLB) is the second most common form of dementia after Alzheimer’s.
The clinical and pathological presentation of DLB shares similarities with both Parkinson’s (movement and autonomic dysfunction) and Alzheimer’s (dementia). In DLB, like in Parkinson’s, the cytoplasmic inclusions (lewy bodies) accumulate in neurons, eventually leading to cell death. The Lewy bodies are comprised of aggregated proteins, primarily aggregated misfolded alpha synuclein. Furthermore, roughly 8% of LBD patients have a mutation in GBA1. In patients with GBA1 mutations, the formation of lewy bodies is potentially being exacerbated by impaired lysosomes leading to the aggregation of alpha synuclein.
Vanqua Bio Therapeutic Hypothesis: Small molecule activators of GCase will prevent the accumulation of Lewy bodies by improving lysosomal function and reducing the accumulation of alpha synuclein.
Vanqua’s Technology Platform
Vanqua Bio’s discovery platform enabled the identification of next-generation GCase activators that can cross the blood-brain barrier for the treatment of GBA-PD, iPD, GBA-DLB and all forms of Gaucher disease. Our patient-derived iPSC platform allows us to study the effects of our activators in disease-relevant models.

Our insights have enabled us to discover novel small-molecule allosteric activators that normalize the activity of GCase and block the accumulation of pathological alpha synuclein in patient-derived dopaminergic neurons.
Complement Pathway
Vanqua Bio’s Scientific Focus: Our second program targets the inflammation that is associated with many peripheral inflammatory & neurodegenerative diseases.
Complement system – C5aR1
The complement system is a powerful regulator of the innate immune system. When the system becomes aberrantly or over-activated, inflammation or cell lysis can damage susceptible cells and organs in the body.
The complement system can be activated via a diverse set of exogenous or endogenous insults that drive one of the three major activation pathways: classical, alternative, and lectin. The three pathways converge at complement C3 and further downstream at complement C5. The terminal component of the complement system, composing of C5, which is then cleaved to C5a and C5b, is a major contributor to the complement systems effector functions. C5a signals through two G protein-couple receptors with opposing functions, C5aR1 and C5aR2. C5a-C5aR1 binding leads to recruitment and activation of white blood cells (including neutrophils) to sites of injury; whereas C5a-C5aR2 binding reduces inflammation and C5a’s signaling potential.

Several components of the complement system have been clinically validated as compelling therapeutic targets across a range of diseases. C5aR1 antagonism is uniquely attractive within the complement system as the safest way to target complement-driven inflammation. By sparing upstream complement component signaling, C5aR1 antagonism preserves the actions of components that are key for pathogen defense (e.g. neisseria) and beneficial removal of damaged cells.
In clinical trials, C5aR1 antagonism has been proven to reduce the use of steroids in a life-threatening renal disease and has potential to improve standard of care in a range of related diseases. In the CNS, human genetics link components of the complement system to multiple neurodegenerative diseases. Aberrant C5aR1 activation in the CNS promotes harmful activation of CNS immune cells, which leads to decreased cell function and increased cell death. Antagonism of C5aR1 in the CNS is an attractive approach to a number of conditions, including Alzheimer’s disease, to reduce neuroinflammation and slow disease progression.
Vanqua Bio Therapeutic hypothesis: small molecule antagonists of C5aR1 will reduce pathogenic complement-mediated inflammation in central diseases

Source: Mastellos et al., Nature Review Drug Discovery (2019); Capriani et al., Frontiers in Immunology (2019)
Related Publications
GCase
Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease
Sidransky E. et al (2009) New England Journal of Medicine
GBA-Associated Parkinson’s Disease and other Synucleinopathies
Gan-Or Z. et al (2018) Current Neurology and Neuroscience Reports
Glucocerebrosidase and its Relevance to Parkinson Disease
Do J. et al. (2019) Molecular Neurodegeneration
Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity
Ysselstein D. et al (2021) Movement Disorders
Glucocerebrosidase activity and lipid levels are related to protein pathologies in Parkinson’s disease
Leyns C. et al (2023) NPJ Parkinson’s Disease
Complement Pathway
Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System
Carpanini S. et al. (2019) Frontiers in Immunology
Clinical promise of next-generation complement therapeutics
Mastellos D. et al. (2019) Nature Reviews Drug Discovery
Complement in neurological disorders and emerging complement-targeted therapeutics
Dalakas M. et al. (2020) Nature Reviews Neurology